Research
My research interests primarily lie in the following fields:
For the complete publication list, please kindly refer to my Google Scholar.
1. Mass Spectrometry Imaging
1.1 Introduction
Mass spectrometry imaging (MSI) is a well-established analytical technique that allows direct mapping of a wide variety of chemical classes from different biological samples. It provides rich information concerning analyte identity, relative abundance, and spatial distribution. MSI has enjoyed a surge in popularity in the last decade due to its non-targeted and label-free nature. Analytes of interest do not have to be pre-selected or even known prior to MSI analysis, and they can in most cases be detected without any chemical modification or labeling. This is in stark contrast to most histochemical staining techniques, which require the availability of a suitable antibody. Most importantly, MSI provides spatio-chemical information which is much more specific as compared to different kinds of microscopic imaging techniques, and much more intuitive similar to colorimetric imaging.
MSI entails many different types of platforms, among which matrix-assisted laser desorption/ionization (MALDI), desorption electrospray ionization (DESI), and secondary ion mass spectrometry (SIMS) are most popular. Despite differences in spatial resolution, chemical scope, and speed, all MSI-based approaches rely on discrete (e.g. MALDI) or continuous (e.g. DESI) movement of a probing beam across a sample surface for spatial analysis (Fig. 1). Molecules present at each measurement coordinate are desorbed and ionized, and a mass spectrum is thus recorded along with its current location (Fig. 1). Several thousand mass spectra are typically acquired in this manner. After measurement, an MS image can be generated by extracting the intensity values of a selected m/z peak from each acquired spectrum. The extracted intensities are then plotted as colour scale values (pseudo-colour) for each pixel in a grid representing the original locations on the sample (Fig. 1). The resulting MS image represents the spatial intensity distribution of an m/z peak, which can be tentatively assigned to a certain compound. It is important to note that separate MS images can be produced for each m/z peak in a single MSI experiment. Therefore, MSI is an untargeted and multiplexed technique. The number of MS images generated in theory equals the number of distinct m/z peaks detected and resolved in the spectra, which can range from tens to thousands. Optionally, an optical image of the very same sample can be co-reregistered with an MS image for detailed inspection of the spatial organization of an analyte of interest. Several different MS images can be also overlaid to compare their distributions on the sample.
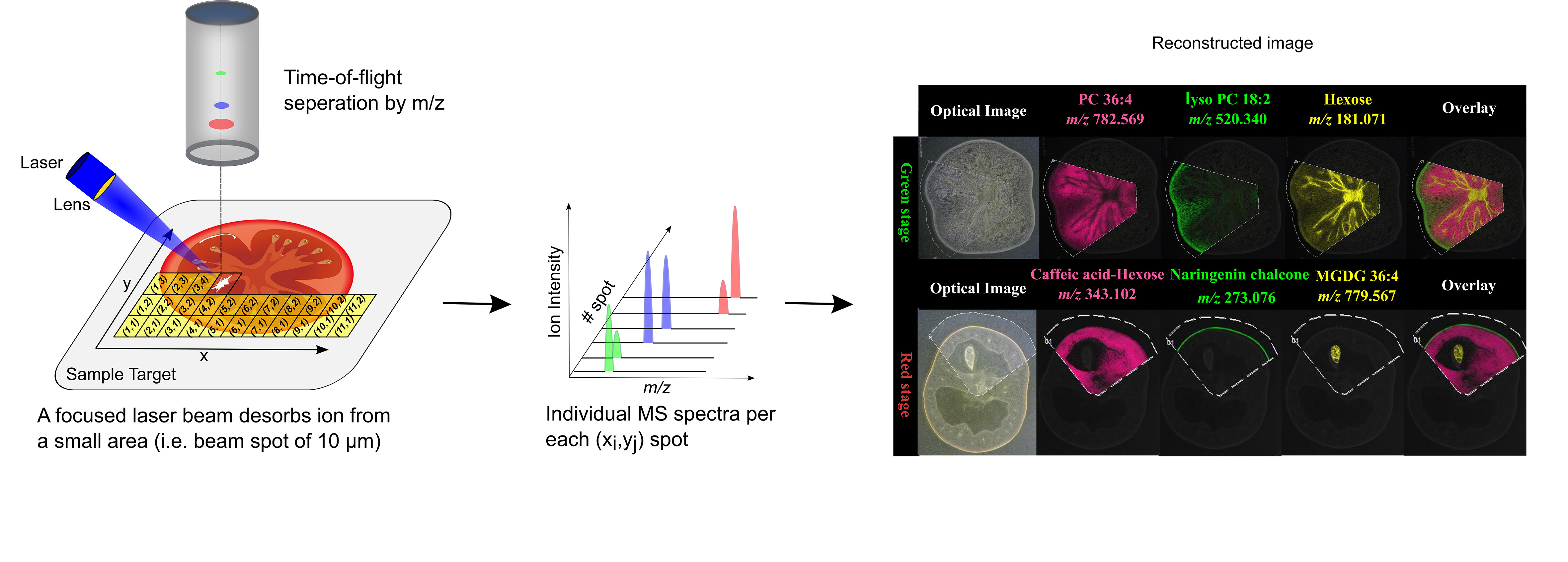
1.2 Representative publications
-
Dong, Y., Shachaf, N., Feldberg, L., Rogachev, I., Heinig, U. and Aharoni, A., 2023. PICA: Pixel Intensity Correlation Analysis for Deconvolution and Metabolite Identification in Mass Spectrometry Imaging. Analytical Chemistry, 95(2), pp.1652–1662. PDF
-
Dong, Y., and Aharoni, A., 2022. Image to insight: exploring natural products through mass spectrometry imaging. Natural Product Reports. PDF
-
Dong, Y., Sonawane, P., Cohen, H., Polturak, G., Feldberg, L., Avivi, S.H., Rogachev, I. and Aharoni, A., 2020. High mass resolution, spatial metabolite mapping enhances the current plant gene and pathway discovery toolbox. New Phytologist, 228(6), pp.1986-2002. PDF
-
Dong, Y.., Li, B. and Aharoni, A., 2016. More than pictures: when MS imaging meets histology. Trends in plant science, 21(8), pp.686-698. PDF
-
Dong, Y., Li, B., Malitsky, S., Rogachev, I., Aharoni, A., Kaftan, F., Svatoš, A. and Franceschi, P., 2016. Sample preparation for mass spectrometry imaging of plant tissues: a review. Frontiers in Plant Science, 7, p.60. PDF
2. Metabolomics
2.1. Introduction
Metabolomics is a field of study that utilizes various analytical tools to quantify and qualitatively analyze small molecules (metabolites) in a given sample. Depending on the research question or specific application, there are three common metabolomics strategies:
Untargeted Assay: (or frequently referred to as untargeted metabolomics or global metabolomics). Its objective is to reproducibly measure as many metabolites as feasible, and provide semi-quantitative data (chromatographic peak areas are reported, not concentrations). The chemical identity of metabolites is not necessarily known prior to data acquisition.
Targeted Assay: (or sometimes referred to as targeted metabolomics). This approach focuses on a small number of metabolites of interest whose chemical identity is known prior to data acquisition, and an absolute concentration of each metabolite is typically reported.
Semi-targeted Assay: (or frequently referred to as metabolic profiling). It acts as an intermediate between untargeted and targeted analyses, where typically hundreds of metabolites are targeted, whose chemical identity is known prior to data acquisition. Semi-quantification information is reported in this type of approach.
2.2 Representative publications
-
Gou, M., Duan. X., Li, J., Wang, Y., Li, Q., Pang, Y., Dong, Y (corresponding author)., 2022. How do Vampires Suck Blood? PDF
-
Arya, G.C., Dong, Y. (co-first author), Heinig, U., Shahaf, N., Kazachkova, Y., Aviv-Sharon, E., Nomberg, G., Marinov, O., Manasherova, E., Aharoni, A. and Cohen, H., 2022. The metabolic and proteomic repertoires of periderm tissue in skin of the reticulated Sikkim cucumber fruit. Horticulture Research, 9. PDF
3. Stable Isotope Labeling
3.1 Introduction
Stable isotopes have the same number of protons as common elements but differ in masses due to difference in the number of neutrons. Therefore, isotopologues (i.e., metabolites containing stable isotopes) and their unlabelled counterparts generally behave identically in LC but can be distinguished in MS by their m/z values. Stable isotope labelling (SIL) is increasingly used in different areas of metabolomics research and it shows great potential in metabolite structural elucidation, quantification, and pathway analysis.
Depending on the labelling regime, organisms can be labelled uniformly or non-uniformly with different degrees of enrichment. In “global SIL” approaches, stable isotopes are introduced into biological systems via main nutrient sources in order to completely label all endogenously produced metabolites; while in “tracer-based SIL”, a labelled compound (namely tracer) is administrated to the organism as a metabolic substrate. Uptake and incorporation of the tracer allow following the metabolic fate of the tracer and its flux through specific pathways.
Together with my colleague, Feldberg Liron, we have extended the single tracer-based SIL approach by simultaneously feeding the organisms with the same precursor metabolite possessing two (now even three) different labeling schemes. Compared to the conventional single stable isotope tracing, this approach, termed DLEMMA (Dual Labeling of Metabolites for Metabolome Analysis), could largely reduce the number of plausible molecular formulas and chemical structures of the detected metabolites, thus facilitating metabolite identification. In addition, we have also successfully coupled DLEMMA with mass spectrometry imaging for accurate spatial localization analysis.
3.2 Representative Publications
-
Dong, Y., Feldberg, L., Rogachev, I. and Aharoni, A., 2021. Characterization of the PRODUCTION of ANTHOCYANIN PIGMENT 1 Arabidopsis dominant mutant using DLEMMA dual isotope labeling approach. Phytochemistry, 186, p.112740. PDF
-
Dong, Y., Feldberg, L. and Aharoni, A., 2019. Miso: an R package for multiple isotope labeling assisted metabolomics data analysis. Bioinformatics, 35(18), pp.3524-3526. PDF
-
Feldberg, L., Dong, Y. (co-first author), Heinig, U., Rogachev, I. and Aharoni, A., 2018. DLEMMA-MS-imaging for identification of spatially localized metabolites and metabolic network map reconstruction. Analytical chemistry, 90(17), pp.10231-10238. PDF
4. Chemoinformatics
4.1 Introduction
Both mass spectrometry imaging (MSI) and metabolomics studies generate increasingly complex data sets. Their comprehensive evaluation requires specialized and efficient data analysis approaches that involves cheminformatics, and statistics.
Apart from routine MSI and metabolomics data analysis, I also actively developing novel data analysis methods and software tools (Fig. 4) in these two research fields. R and Python are my commonly used programming languages.
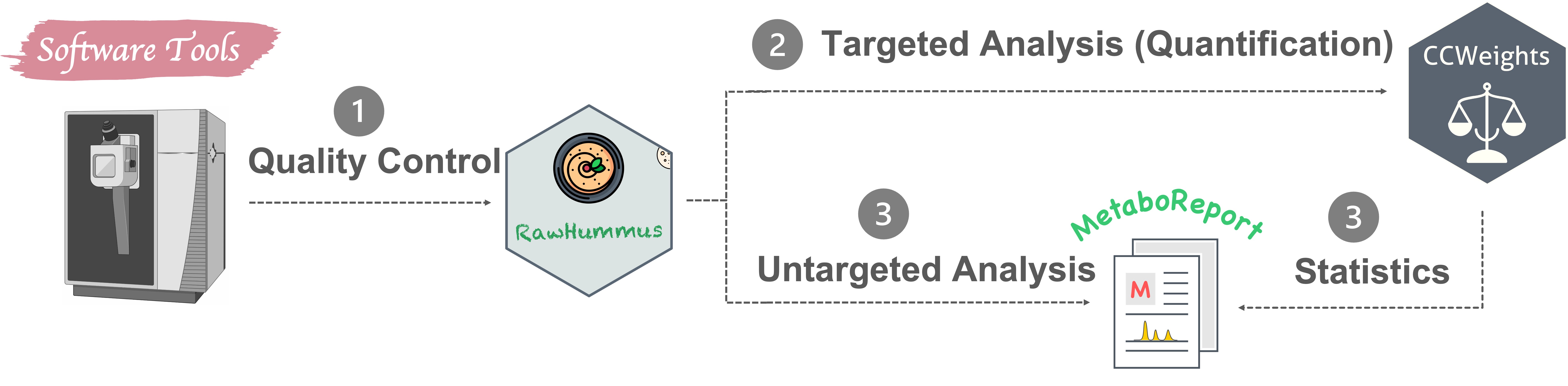
4.2 Software I developed
-
MSbox : A series of common mass spectrometry tools. It allows checking element isotopes, calculating (isotope labelled) exact monoisitopic mass, m/z values and mass accuracy, and inspecting possible contaminant mass peaks, examining possible adducts in electrospray ionization (ESI) and Matrix-Assisted Laser Desorption Ionization (MALDI) ion sources. GitHub
-
Miso: An efficient tool for fishing out labeled molecules from single, dual or multiple isotope labeling experiment. GitHub
-
CCWeights: The accuracy of any analytical method highly depends on the selection of an appropriate calibration model. CCWeights is designed to automatically assess & select the best weighting factors (WF) for accurate metabolite quantification using the linear calibration curve. GitHub
-
RawHummus: Robust and reproducible data is essential to ensure high-quality results for metabolomics studies where detector sensitivity drifts, retention time, and mass accuracy shifts frequently occur. RawHummus is designed to automatically detect measurement bias & verify system consistency. GitHub
-
MetaboReport: Provides a flexible and user-friendly workflow for data cleaning, pre-processing, statistical analysis, and reporting of metabolomics, lipidomics, and proteomics. (not published yet)
4.3 Representative publications
-
Dong, Y., Kazachkova, Y., Gou, M., Morgan, L., Wachsman, T., Gazit, E. and Birkler, R.I.D., 2022. RawHummus: an R Shiny app for automated raw data quality control in metabolomics. Bioinformatics, 38(7), pp.2072-2074. PDF
-
Dong, Y., Wachsman, T., Morgan, L., Gazit, E. and Birkler, R.I.D., 2021. CCWeights: an R package and web application for automated evaluation and selection of weighting factors for accurate quantification using linear calibration curve. Bioinformatics Advances, 1(1), p.vbab029. PDF
-
Dong, Y., Feldberg, L. and Aharoni, A., 2019. Miso: an R package for multiple isotope labeling assisted metabolomics data analysis. Bioinformatics, 35(18), pp.3524-3526. PDF